BiasAway web-server¶
Introduction¶
The BiasAway web-server provides an interactive and easy to use interface for users to upload FASTA files and to generate background sequences. It comes with precomputed genomic partitions of 100, 250, 500, 750, and 1000 bp bins for the genome of nine species (Arabidopsis thaliana; Caenorhabditis elegans; Danio rerio; Drosophila melanogaster; Homo sapiens; Mus musculus; Rattus norvegicus; Saccharomyces cerevisiae; and Schizosaccharomyces pombe). These background sequences are provided through Zenodo at 10.5281/zenodo.3923866. These background sequences were generated using the script at https://bitbucket.org/CBGR/biasaway_background_construction, which can be used by users to generate their own background sequences. The result page provides information about mononucleotide, dinucleotide, and length distributions for the provided and generated sequences for comparison.
BiasAway has four modules:
Note
The BiasAway web-application automatically generate distribution plots for QC. Plots provide information about distribution of %GC, dinucleotides, and lengths for the input sequences and generated sequences. Moreover, BiasAway provides the following QC metrics for comparing these distributions whenever possible: mean absolute error and goodness of fit computed as Pearson’s chi-squared statistic, log-likelihood ratio test (G-test), and the Cressie-Read power divergence.
Below are screenshots for individual modules.
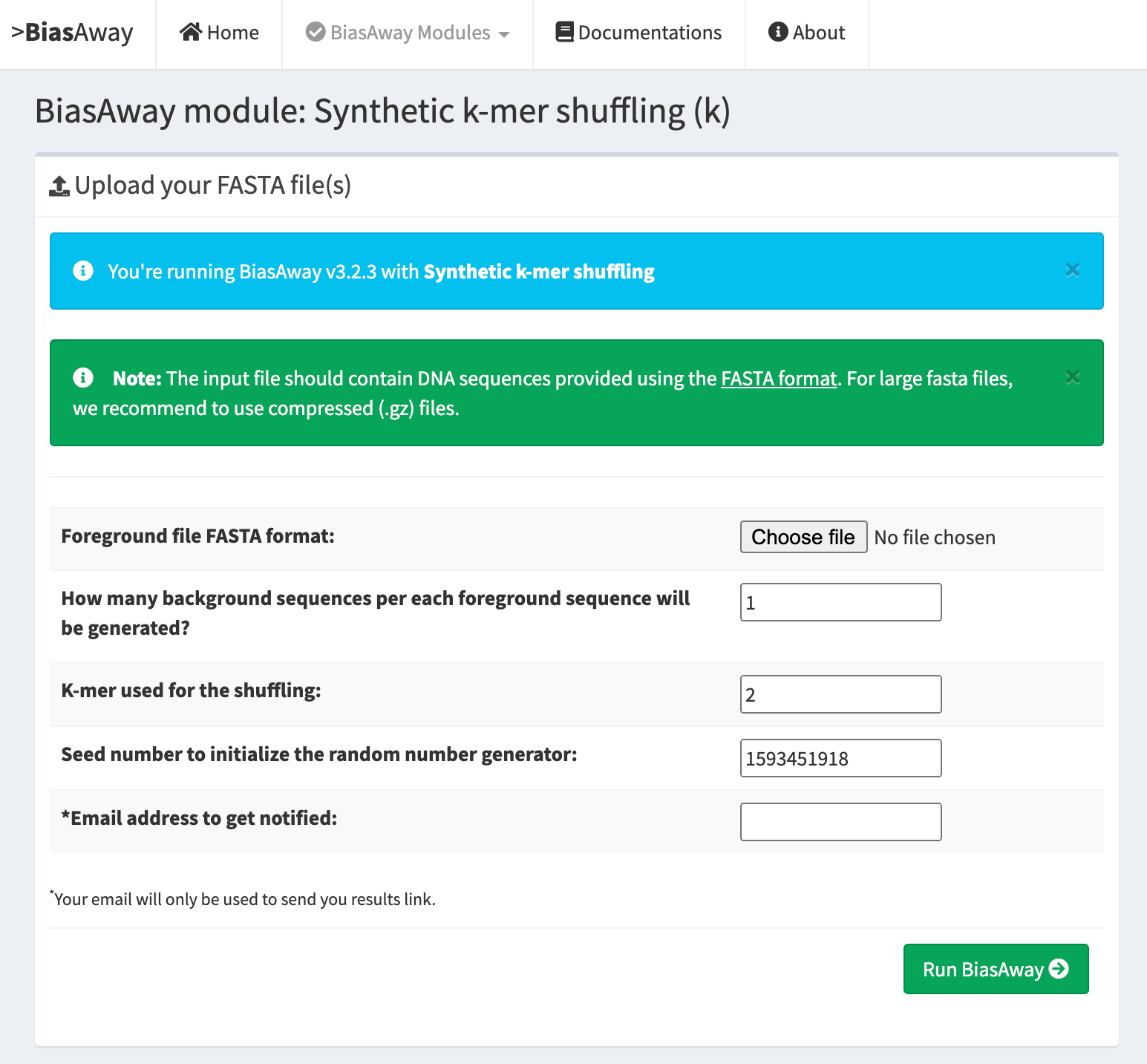
K-mer shuffling¶
This module should be run when the user aims at preserving the global k-mer nucleotide frequencies of input sequences.
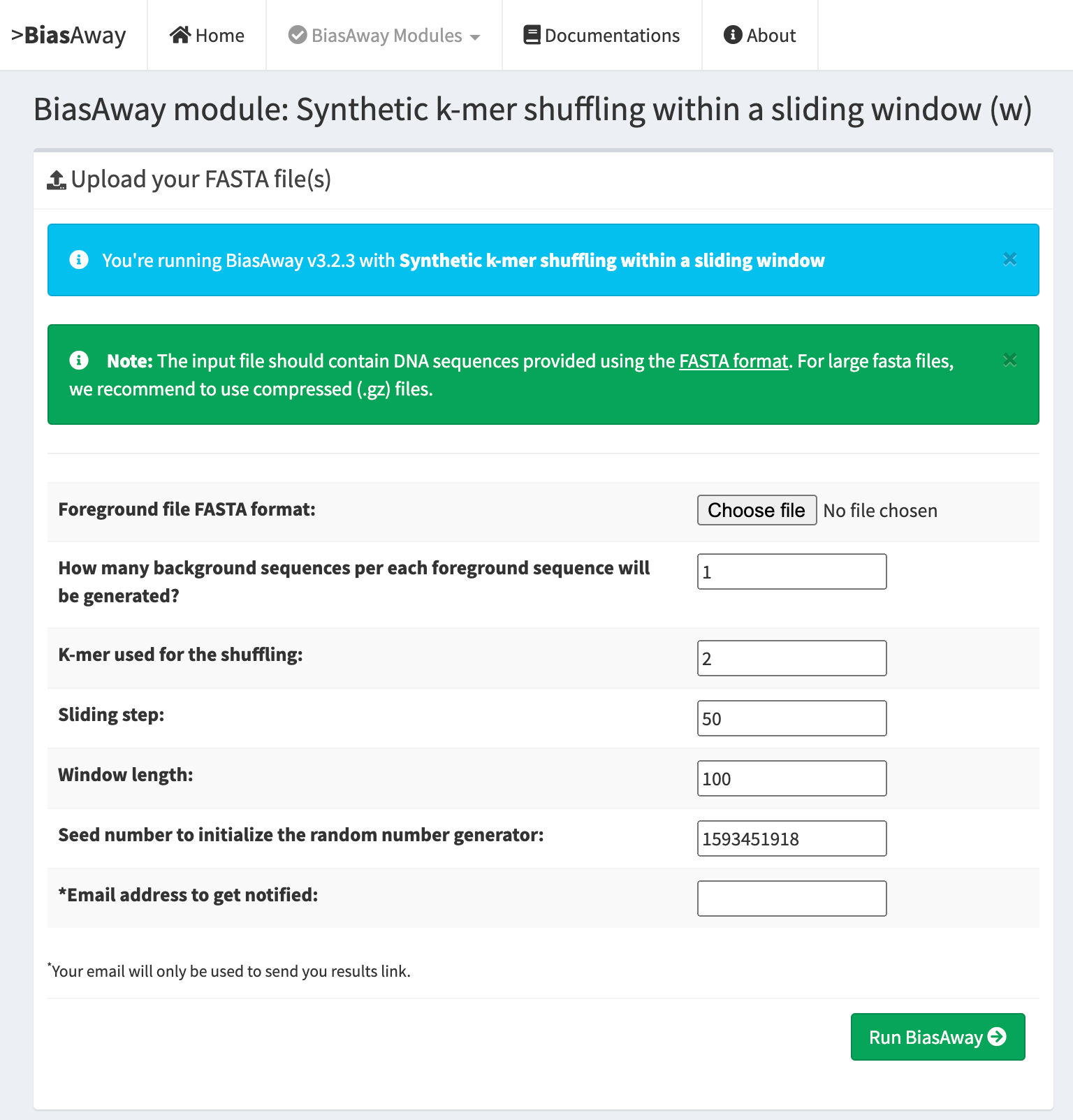
K-mer shuffling within a sliding window¶
This module should be run when the user aims at preserving the local k-mer nucleotide frequencies of input sequences.
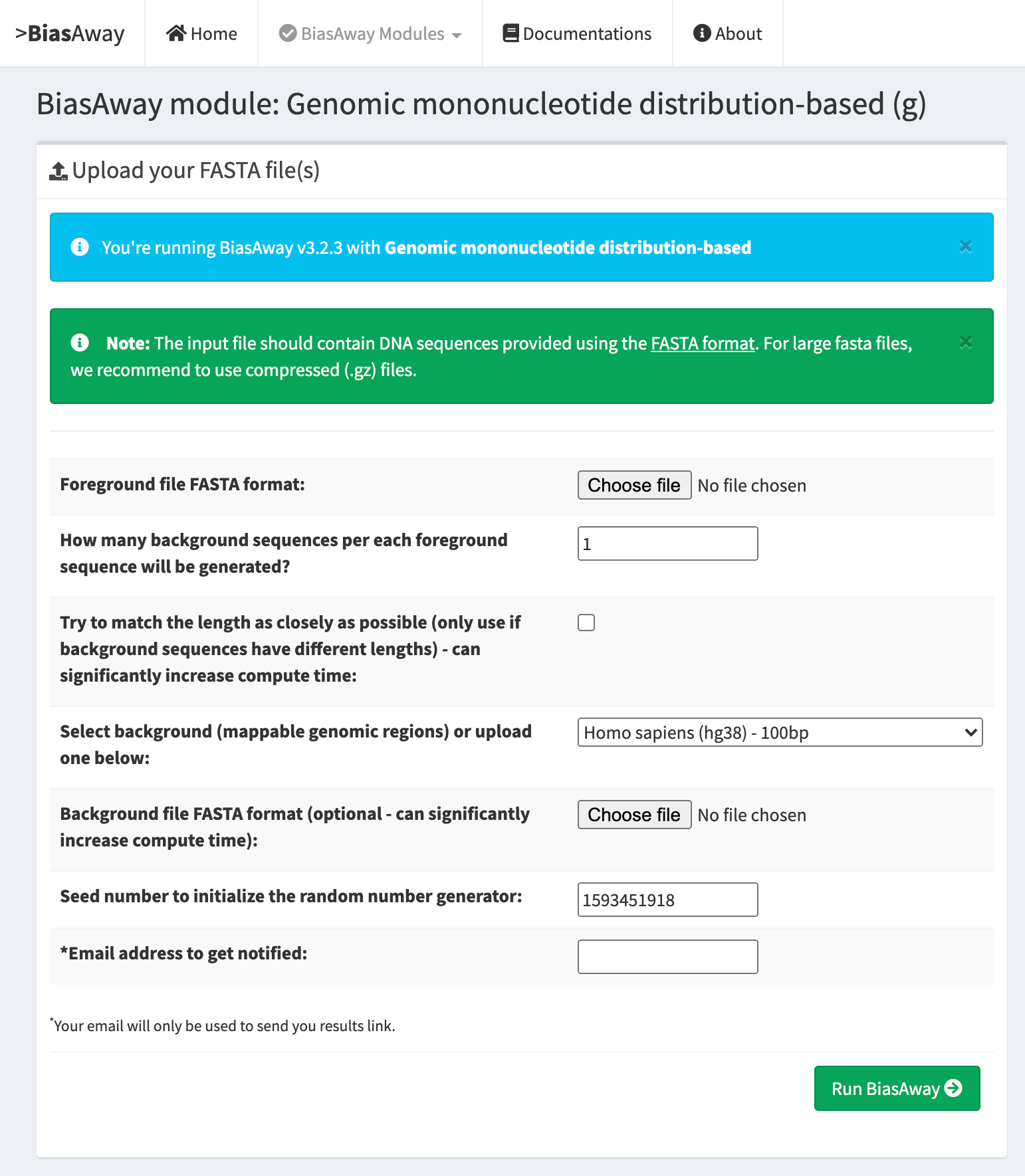
Genomic mononucleotide distribution matched¶
This module should be run when the user aims at selecting genuine genomic background sequences from a pool of provided genomic sequences to match the distribution of mononucleotide for each target sequence.
Genomic mononucleotide distribution within a sliding window matched¶
This module should be run when the user aims at selecting genuine genomic background sequences from a pool of provided genomic sequences to match the local distribution of mononucleotide for each target sequence.
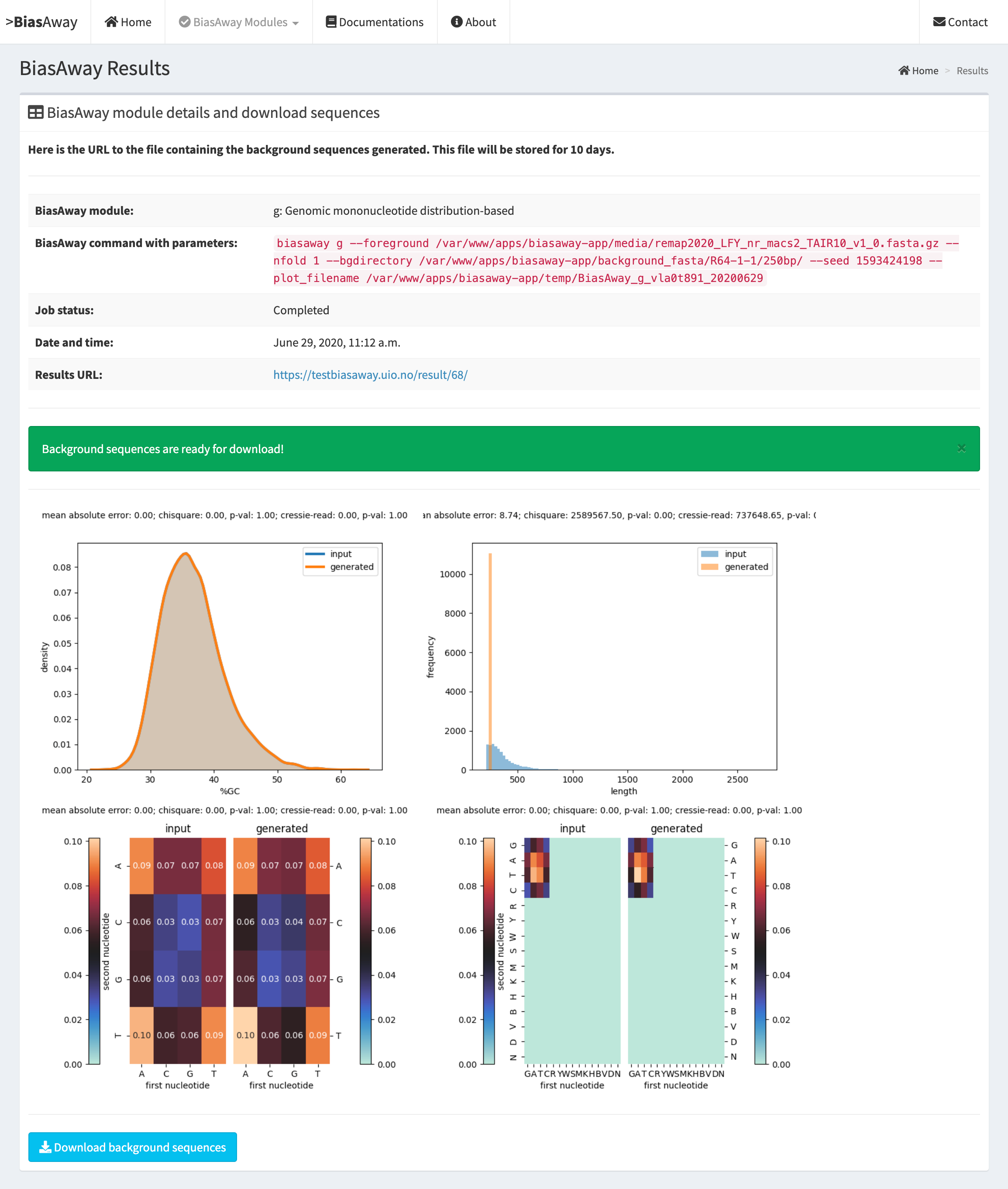
Example result page and QC plots¶
BiasAway provides quality control (QC) plots and metrics to assess the similarity of the mono- and di-nucleotide, and length distributions for the foreground and background sequences. Specifically, four plots are provided to visualize how similar the foreground and background sequences are when considering (2) their distributions of %GC content using density plots, (2) their dinucleotide contents considering all IUPAC nucleotides using a heatmap, (3) their dinucleotide contents considering adenine, cytosine, guanine, and thymine nucleotides using a heatmap, and (4) their distributions of lengths.
Generation of background repositories¶
Modules g and c of BiasAway require the generation of a background repository for the genome of interest. This can be created with the script located at our BitBucket repository.
Our BiasAway Web-Server contains precomputed background repositories for 9 species. The genome fasta files used to create these can be found below:
- Homo sapiens: GRCh38/hg38
- Mus musculus: mm10
- Rattus norvegicus: Rnor 6.0
- Arabidopsis thaliana: TAIR10
- Danio rerio: GRCz11
- Drosophila melanogaster: dm6
- Caenorhabditis elegans: WBcel235
- Saccharomyces cerevisiae
- Schizosaccharomyces pombe: ASM294v2
Please note that some genome fasta files are separated by chromosomes in their original repositories. In that case, please make sure to concatenate all chromosome fasta files in one single genome fasta file.
We also provide a collection of precomputed background repositories for the nine organisms mentioned above using k-mers of size 100, 250, 500, 750 and 1000 base pairs. They can be found as individual compressed files in our Zenodo repository